锂硫电池的高能量密度使其成为下一代储能技术的有力候选者,其正负极的主要活性物质分别是硫和锂,在充放电过程中会形成可溶的高阶多硫化物从正极扩散至负极,导致正极活性物质的流失以及负极的失活,即所谓的“穿梭效应”。同时,正极处缓慢的硫还原动力学也限制了锂硫电池的性能。近年来,有关研究发现应用在锂硫电池正极材料中的负载型单原子催化剂可以抑制“穿梭效应”以及加快硫还原反应动力学。然而,单原子催化剂的结构种类繁多,超出了传统的“试错法”或常规DFT计算的能力,急需进行系统研究。
近日,金属所沈阳材料科学国家研究中心联合研究部李波副研究员的研究小组,采用基于高通量密度泛函理论计算的机器学习方法,系统研究了多硫化物的吸附模式,并对上千种氮掺杂碳材料负载的过渡金属单原子催化剂进行了筛选,为锂硫电池正极材料中单原子催化剂的设计提供了指导。这一成果近日发表于The Journal of Physical Chemistry Letters(doi: 10.1021/acs.jpclett.1c00927)上,并被选为封面文章(cover article)。同时ACS和J. Phys. Chem. 推特分别对该工作进行了介绍和评论。
研究人员首先采用密度泛函理论计算对800余个吸附结构进行计算,结果显示多硫化锂在催化剂上有四种可能的吸附构型,并可分为两大类,即解离吸附和非解离吸附。基于晶体图卷积神经网络训练的分类器,成功地区分了发生S-S键断裂的吸附与其他类型的吸附。研究人员进一步对吸附构型的电子结构分析,显示负载金属原子后催化剂与多硫化锂间的相互作用发生明显变化,因而使吸附显著增强,从而减少“穿梭”过程的发生。此外,机器学习训练出的回归模型对吸附能也具有较好的预测能力,其平均绝对误差为0.14 eV。基于这一模型,预测了上千个吸附构型的吸附能,利用过电势的计算给出了相应的火山型曲线。同时,结合可溶性多硫化物的吸附能,预测并筛选出了数个性能均衡的单原子催化剂。目前的工作不仅拓宽了单原子催化剂的应用范围,也为锂硫电池正极材料的设计提供了新思路和新方向。
该工作得到了国家自然科学基金、辽宁省自然科学基金材料联合基金、NSFC-广东联合基金(第二期)超级计算科学应用研究专项等资助。
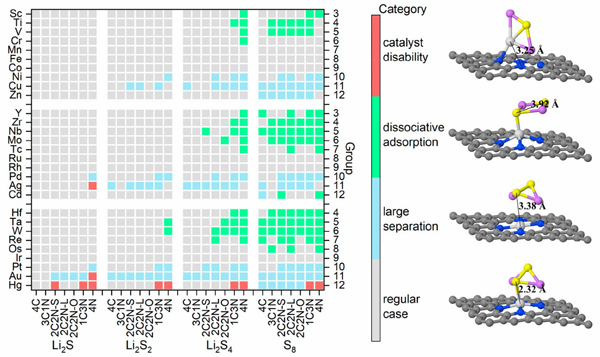
图1. 四种可能吸附构型的分布及结构图示。
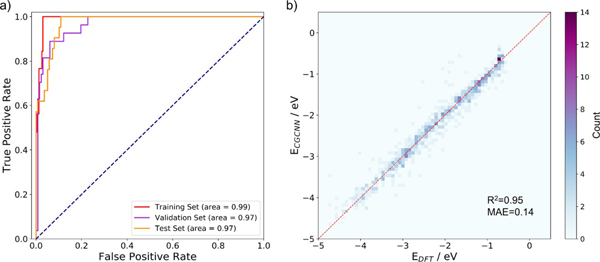
图2. 机器学习模型的性能。a) 分类性能,b) 回归性能。
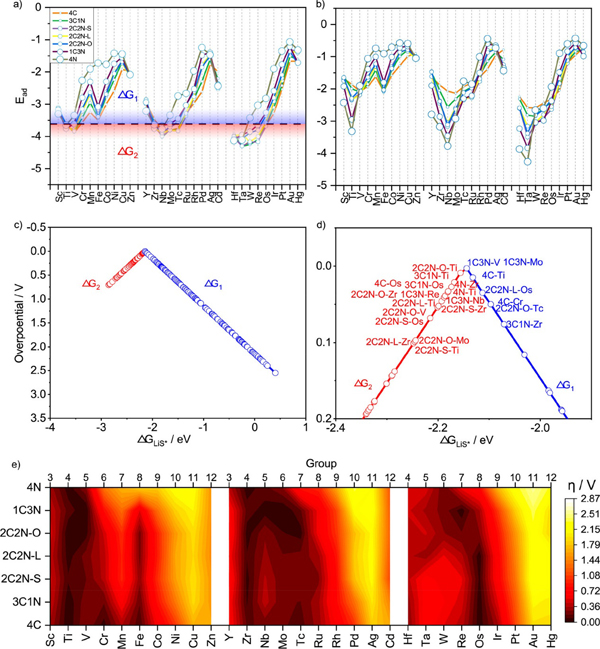
图3. 多硫化锂吸附能及过电势的预测。a) LiS*的吸附能,b) 不同催化剂上最弱的可溶性多硫化锂吸附能,c)所预测的全部催化剂的“火山图”,d) 过电势低于0.1V的催化剂,e) 预测的过电势。
图4. 从左到右:封面图片、 Journal of Physical Chemistry 推特介绍、ACS推特介绍。
 官方公众号
官方公众号
