低碳烯烃是化工产业的支柱,是合成塑料、橡胶和纤维的基本原材料。烯烃产量是衡量一个国家化工产业能力重要指标之一,随着经济发展烯烃需求在持续增加,2020年我国烯烃消耗量占全球15%以上。由此可见,进一步提高烯烃生产效率有着重要经济价值和社会意义。另一方面,通过烷烃催化脱氢反应可以高效将低碳烷烃分子转化为同碳烯烃。目前,烷烃催化脱氢(直接和氧化)反应面临着选择性低、积碳以及高能耗等挑战,设计和开发新型高效催化剂是解决这些问题的关键。在分子水平上揭示催化剂性质和反应机理是进行有的放矢筛选高效催化剂的前提条件。2014年以来,联合研究部李波副研究员的研究小组一直从事低碳烷烃催化脱氢反应的第一性原理计算模拟研究工作。基于计算结果,揭示了碳基催化剂sp2/sp3核壳结构在脱氢反应中的独特催化性能(ACS Catalysis,7,3779-3785 (2017)),给出了工业铂基催化剂积碳过程的详细描述以及控制积碳的方法(ACS Catalysis, 8, 4694-4704 (2018))。
近日,联合研究部李波研究小组采用基于第一性原理的微观反应动力学模拟方法,系统研究了纳米碳催化乙苯氧化脱氢的反应机理。研究结果表明反应主要通过ER机理发生,而自由基反应在特定条件下也起到了重要作用。相关成果为未来新型非金属碳基催化剂的理性设计及反应条件的优化提供了指导。这一成果近日发表在国际催化知名期刊ACS Catalysis上(doi:10.1021/acscatal.0c02952)。
该工作结合第一性原理计算与微观动力学模拟研究了纳米碳催化剂上乙苯氧化脱氢反应在不同反应条件下的反应机理及其变化。考虑了活性位点再生的三种可能的机理。此外,创新性地发现了弱吸附物种与自由基之间的反应过程。从第一性原理计算中获得基元反应的反应能和势垒,并将其做为微观动力学模拟的输入。第一性原理计算和微观动力学分析表明,酮羰基和弱吸附的HO2·自由基均具有催化烷烃脱氢的活性。ER机理比LH和MvK机理更有可能发生。此外,在特定条件下反应也可以通过弱吸附HO2·发生。在接近化学计量比时,乙苯和氧气反应级数分别为一级和零级。反应速率与乙苯分压呈线性关系,而不随氧气分压变化。催化剂中的缺陷位点通常被认为是高活性位点,计算结果也证实了其高活性,但初始催化剂中缺陷位点密度不影响催化剂本征活性及稳态的反应状态。当前工作不仅加深了对纳米碳上烷烃氧化脱氢机理的理解,而且指认了非金属催化剂的独特性质。
该工作得到了国家自然科学基金、辽宁省自然科学基金材料联合基金、NSFC-广东联合基金(第二期)超级计算科学应用研究专项等资助。
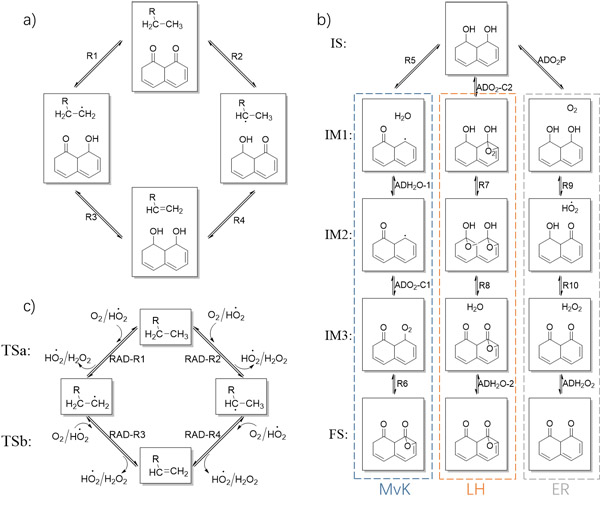
图一、纳米碳催化剂上乙苯氧化脱氢反应网络。a)脱氢反应,b)催化剂活化反应,c)自由基反应
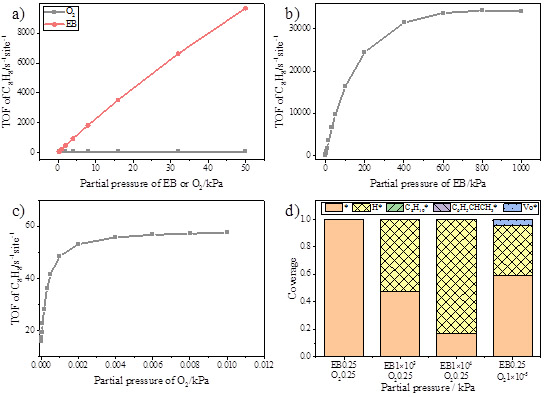
图二、不同条件下苯乙烯的生成速率以及相关主要物种的分布。a) 乙苯或氧气分压为0.25kPa时,另一气体分压在0到50kPa变化时反应的速率,b) 氧气分压为0.25kPa时,乙苯分压在0到1000kPa变化时反应的速率,c) 乙苯分压为0.25kPa时,氧气分压在0到0.01kPa变化时反应的速率,d) 不同分压下主要的吸附物种分布
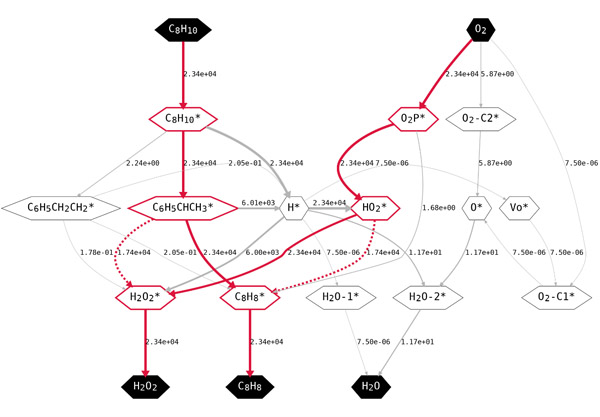
图三、反应网络及800 K, 1 bar EB, 1 bar O2条件下的物种转换速率。红色高亮部分为关键中间物种与反应路径,其中虚线表示自由基反应路径,线右侧的数字为物种转换的速率
 官方公众号
官方公众号
